 手機版
手機版 化工儀器網手機版
化工儀器網手機版
 化工儀器網小程序
化工儀器網小程序
 官方微信
官方微信 公眾號:chem17
公眾號:chem17
 掃碼關注視頻號
掃碼關注視頻號












薄層色譜成像分析儀中不同溶劑對樣品的洗脫能力不同,因此在點樣的同時,樣品在原點開始呈環形展開,如果樣品在溶劑中的溶解度很大,原點將變成空心環,Kaiser稱這種現象為“點樣環形色譜效應”。這種效應對隨后的線形展開造成不良影響,因此配制樣品溶液時應選用對組分溶解度相對較小的溶劑;溶劑的粘度不宜過高,以便于點樣,溶劑的沸點要適中,沸點過低由于揮發會改變樣品溶液濃度導致較大誤差,沸點過高,樣品溶液的溶劑會在原點殘留,導致改變展開劑的選擇性,特別是樣品溶液的溶劑與展開劑的極性相差較大時更為明顯,zui常用的溶劑為甲醇及乙醇,有時也用丙酮。點樣后必須將溶劑全部除去后再進行展開,但要避免高溫加熱,以免改變待測成分的性質。
薄層色譜成像分析儀點樣方式、點樣量及點樣設備的選擇決定于分析的目的、樣品溶液的濃度及被測物質的檢出靈敏度。
點樣體積經典薄層一般為1-5μl,薄層為100-500nl,樣品溶液濃度一般在0.01%-1.00%范圍內,點樣過多會造成原點“超載”,展開劑產生“繞行”現象使斑點拖尾或重疊,使掃描峰形不對稱或不能基線分離,并由于超載得到非線性的校正曲線,嚴重影響定量結果,降低準確度。
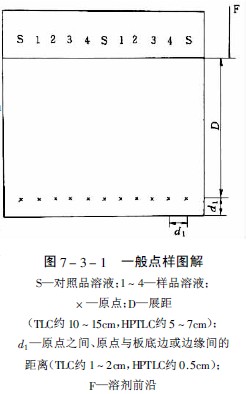
經典薄層的原點直徑zui大不得超過5mm,一般3mm較為合適,薄層原點直徑約為1-2mm,盡可能避免多次點樣,原點直徑過大會降低分辨率及分離度。
經典薄層色譜成像分析儀中薄層起始線距底邊約1.5cm,薄層為1cm,展距前者為10-15cm,后者為5-7cm,對于難分離的化合物用多元展開劑展開時,注意原點與展開劑的距離每次要保持一致,否則由于色譜分離zui初行為的差異而導致分離失敗。
點與點的間距經典薄層為1-2cm,薄層為0.5cm,用具自動換行功能的薄層掃描儀時,斑點之間的距離必須十分,才能正確地掃描每行的色斑,得到好的測定結果,圖7-3-1為一般的點樣圖解。
在點樣前,應將薄層板在日光及紫外光下檢查板面有無損壞或污染,選擇合格的薄層板后決定用點狀點樣還是用帶狀點樣。為此,瑞士CAMAG公司生產了系列點樣設備以供選用,這些設備只適用于硬板。下面是常用的點樣方式及設備。 北京威鋒技術開發公司
(一)點狀點樣
如果薄層色譜成像分析儀僅僅為了定性,用內徑約0.5mm管口平整的毛細管或微量注射器將樣品溶液點在距薄層底邊約2cm處,點樣直徑不超過5mm,點間距約為1-1.5cm即可。
為進行定量,借毛細作用吸樣的定容管有兩種,一是容積為0.5、1、2、3、4及5μl的定量毛細管(Microcaps),另一種是100及200nl的鉑銥合金定量毛細管(Nanopipette);注射器式的可變體積的點樣器有50-230nl的毫微點樣器(Nano-Applicator)及0.5-2.3μl的微量點樣器(Micro-Applicator),用于需要調節體積及沒有毛細作用的鍵合相薄層的點樣。
毛細管式的微量點樣器裝在手動的Nanomat型點樣裝置的磁性點樣頭上,點樣頭裝在彈簧上。按下此點樣頭,毛細管借恒定的壓力接觸薄層表面。點間距離由帶準確的#44 間隔的E字形缺刻裝置控制,適用于普通或薄層板,點樣體積,樣品原點小,原點間隔定位準確;注射器式的毫微點樣器及微量點樣器由測微尺控制,也可與Nanomat型點樣裝置配合,以使點樣位置。
電動點樣裝置*適用于上述各種點樣器的配套使用,并且還可以自動控制點樣次數,點樣器在薄居上的停留時間,以及點樣器接觸薄層的速度。還可避免手持毛細管點樣時的用力不勻,由于靠機械控制接觸板面,壓力恒定。此外,還有可以用于環形展開及向心展開的薄層點樣裝置。
(二)帶狀點樣
當樣品溶液體積大、濃度稀時,采用自動點樣設備進行帶狀點樣。定量分析的點樣范圍為1-99μl,制備型分離點樣范圍為5-490μl,作定量標準曲線時,可由同一標準溶液,自動點上不同體積的標準液,故快速簡便,利用內裝的微處理器編序操作,簡單易行。使用時樣品溶液吸在微量注射器中,點樣器不接觸薄層,而是用氮氣將注射器針尖的溶液吹落在薄層上,薄層板在針頭下定速移動點成0-199mm的窄帶。帶狀點樣展開后的斑點不僅分辨率明顯高于點狀點樣,而且精密準確,為定量分析提供*條件。
(三)自動點樣
全自動點樣裝置結合了現代的電子及機械技術,能進行點狀或帶狀點樣,采用先進計算機編程控制,靈活多用,點樣量為10nl-50μl,可隨意設定點樣規范,可應用于單向、雙向、環形或向心色譜展開,高度自動化的點樣使定量分析結果準確。點樣參數可與Scanner Ⅱ掃描儀的CATS掃描分析軟件連用,令定量分析工作達到高度自動化。
(四)接觸點樣
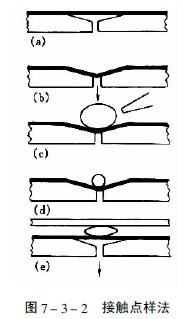
當樣品溶液粘度較大或點樣體積要超過100μl時,用毛細管點樣就有困難,Fenimore設計的接觸點樣法適用于這類樣品的點樣,美國制造的Clarke Contact Spotter就是這種接觸點樣器,見圖7-3-2。在帶有抽氣孔的凹形塊的上方覆蓋一片涂有疏水性物質的聚氟化物薄膜(a),當減壓抽氣時薄膜凹陷(b),此時將樣品溶液點在凹槽處(c),然后用氮氣吹去大部分溶劑(d),再將濃縮后的樣品液滴與薄層板的吸附劑層接觸(e),此時樣品就定量地轉移到薄層上。
北京威鋒技術開發公司
1988年Kaiser對以上各種點樣方式進行了比較,總結其優缺點見表7-3-1、圖7-3-3。
表7-3-1 各種點樣方式的比較北京威鋒技術開發公司
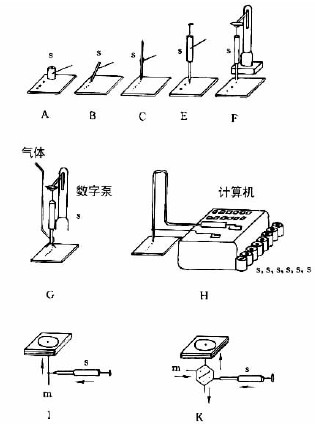
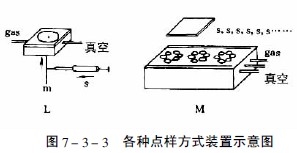
圖7-3-3中A-F為zui常用的毛細管或注射器點樣:A—一次性數毫米長的薄壁玻管;B—長5-60mm,內徑為0.2-0.05mm的熔融石英管,用機械手操作;C—封在玻璃管中的鉑銥合金管(100nl,200nl),可點樣,生物樣品點樣后可用火焰清潔,見圖7-3-4;E—微量注射器,機械控制,接觸板面點樣;F—同E,用步進馬達控制操作;G—同E,樣品溶液用氣體噴到薄層板上;H—同E,*用計算機控制點樣;I、K、L—借泵系統點樣,用于高壓平面液體色譜(HPPLC);M—為Fenimore的固態樣品環形轉移器。
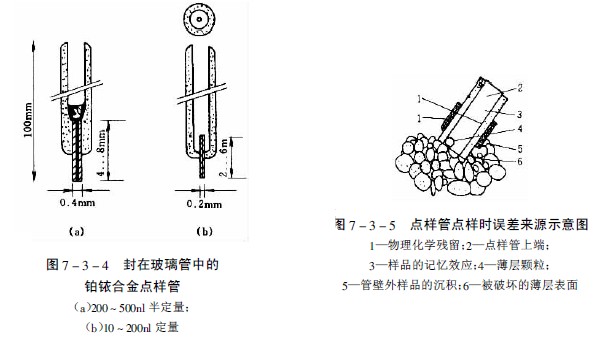
用管狀點樣器上樣的誤差來源是由于點樣時薄層表面的損傷,溶液“爬壁”效應使外壁樣品的沉積、樣品的記憶效應以及管中的微小氣泡等,見圖7-3-5。
點樣誤差的大小與樣品的溶解度,溶液的表面張力,溶劑的粘度、揮發性和極性、點樣所耗費的時間、薄層板的質量(粒度、均勻度、薄層厚度、粘合劑的性質)等有關。
(五)特殊的點樣技術
在進行植物成分薄層分離之前,提取是首要的步驟,常用適當溶劑進行待測物質的提取,但此法操作麻煩、樣品需要量較大,濃縮提取液時對熱不穩定的成分易破壞,因此人們研究了樣品不經提取而直接點樣的方法。
1.熱微量抽出法
半個世紀前,人們將一些藥物進行微量升華,對升華物進行晶型、熔點以及理化性質等鑒定。在zui簡單的情況下,只需將幾毫克生藥粉末放在微量載玻片上加熱,升華物被收集到距離放樣品載玻片1mm的另一塊載玻片上。1968年Stahl將熱微量分離轉移技術與薄層色譜法連用,稱為“熱微量抽出法”此法的優點是大大縮短了提取生藥中揮發性成分前處理的時間,簡化了提取方法,減少了樣品用量,缺點是不宜用于非揮發性成分。
將植物粉末樣品放在一端拉細的玻璃管中,并同時放入含有一定水分的合適添加劑,使加熱過程中保持連續的水蒸氣流(在TAS法中除用含水的分子篩作中性發氣劑外,還可用酸性發氣劑,如丙二酸,在180℃時產生二氧化碳和乙酸,能使樣品中的堿性揮發性成分以鹽的形式保留而不逸出,而酸性揮發性成分隨酸性氣體轉移到薄層上;反之亦可用堿性發氣劑,如含氨的氯化鈣,使樣品中的堿性成分隨氨氣轉移到薄層上),管的一端用硅橡膠薄膜嚴密封閉,并放置在一定溫度的TAS爐上,裝置見圖7-3-6。
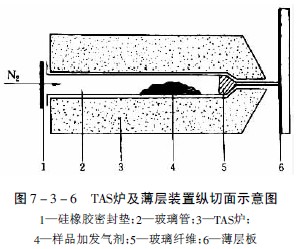
加熱至220℃,揮發性成分氣化,并從玻璃管一端的毛細管口逸出,管中同時以30ml/min的氮氣流幫助揮發性成分逸出,逸出的氣化成分點加到離出口處1mm的薄層上,待上樣完畢,按常規方法展開,即可獲得熱餾分色譜圖。
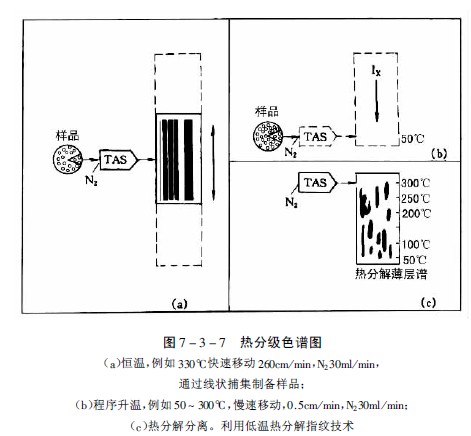
Stahl和Lott發展了此法,設計了可以一定速度橫向移動的薄層板裝置,可使受熱后揮發性成分或熱分解產物附著在移動的薄層板起始線上,加熱方式可以恒溫或程序升溫,從而可以得到各種樣品的特征譜,改進后的方法稱為熱分級色譜法。圖7-3-7為用市售的“TASOMAT”裝置得到的特征圖譜。
這種微量快速的上樣方法,對天然化合物及合成高分子化合物的分離和鑒定已成為一個重要的手段。下村等在中藥厚撲鑒別的研究中應用了此法,只需少量樣品即可鑒別厚撲酚(honokiol);橋本庸平等用此法分離了的旋光異構體,管內殘渣經生物堿檢查呈陰性,證明加熱后的揮發是*的。Jolliffe等將含有某些生物堿的藥物,如烏頭、檳榔、顛茄、金雞納、麥角、曼陀羅、天仙子、蘿芙木、番木鱉等31種植物藥進行熱微量抽出法,逸出的揮發性成分直接點在薄層上,用五種不同展開劑展開,用羅丹明B及奶油黃為參比物,顯色后對所分離的生堿進行鑒定。Cheml等將金雞納樹皮中的生堿在爐溫300℃時揮發至硅膠板上,展開后鑒定辛可寧等七種生堿。Van Meer等成功地用本法分離了纈草根中的纈草素及異纈草素。Liptak等用本法檢驗洋茴香、洋甘菊和歐薄荷以及它們的精油類成分。
中成藥組成復雜,藥典收載的中成藥薄層色譜鑒定法均系用溶劑提取后和單味藥材或單一成分進行對照。曾美怡等試用熱微量轉移法鑒定中成藥,方法簡述如下:
成方制劑的處理:蜜丸拌以等量硅膠5;片劑研成細末;膠囊劑和散劑不加處理;油劑可點在硅膠板上,然后將硅膠連油刮下。
熱微量轉移條件將樣品裝入用鋁箔卷成的細筒,加一粒含水分子篩,置加入管內,于220-225℃加熱60-90s。樣品中的揮發性、熱分解或升華性成分即直接轉移至薄層上。
定位用茴香醛試劑或用熒光法。
樣品用量成方制劑為10-16mg,單味藥為4-8mg。受試樣品有冠心蘇合丸、復方丹參片、七厘散、銀翹解毒丸、六味地黃丸等成方及單味藥,進行了成功的探索,其優點是不需經過復雜的前處理即可得到可供鑒別的色譜。此外,本法也可以應用于小量生藥的半定量分析,生藥成分的微量制備以及生藥的指紋鑒定等。
2.流體提取法
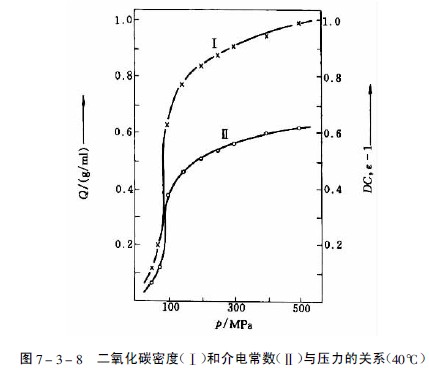
在臨界壓力和臨界溫度以上相區內的氣體為超臨界流體,此時氣體的密度、介電常數和溶劑樣性質均顯著增加,見圖7-3-8。物質在超臨界流體中的溶解度由于壓縮氣體與溶質分子間相互作用加強而大大增加。
改變溫度和壓力就可改變超臨界流體的特性,因此可以分別在一定溫度和壓力條件下將不同極性的成分從樣品中提取出來,這種方法與薄層色譜聯用,稱流體提取一薄層色譜法(Fluid extraction-TLC,FE-TLC)。
用于這種提取過程的氣體有二氧化碳、氧化亞氮、乙烯、三氟甲烷、六氟化碳、氮氣、氬氣等。表7-3-2中列出了各種氣體在臨界點的物理參數。其中二氧化碳是zui常用的一種氣體。
表7-3-2幾種氣體的物理參數
北京威鋒技術開發公司
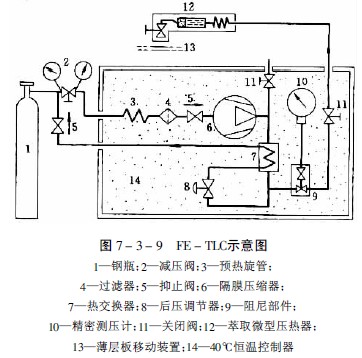
圖7-3-9為FE-TLC示意圖,從高壓氣瓶1出來的氣體,通過電動隔膜壓縮器6壓至7.0-40MPa,壓縮器和微量萃取器12在恒溫器內保持恒溫。萃取器的體積為2ml,被萃取樣品置在其中。先關閉閥門11,待高壓提取器內氣壓達到提取壓力,提取過程達到平衡后即打開。超臨界流體通過一毛細管噴出。由于氣流壓力下降至大氣壓,帶出的提取物析離并吸附在薄層板13上,薄層板與毛細管出口間距離為1-5mm。點樣后按薄層常規方法操作。通過分步提高提取壓力,可以分步提出不同成分。
Stahl以二氧化碳為提取氣體,研究了不同化合物的可提取性與化學結構的關系。實驗結果說明,烴類和親脂性化合物,如酯、醚、內酯和氧化物等,氣壓達7.0-10.0MPa即可提取,結構中引入—OH或—COOH基,則提取困難,對于多羥基酚、多酚基酸、糖和氨基酸等在500個atm下也不能提出。
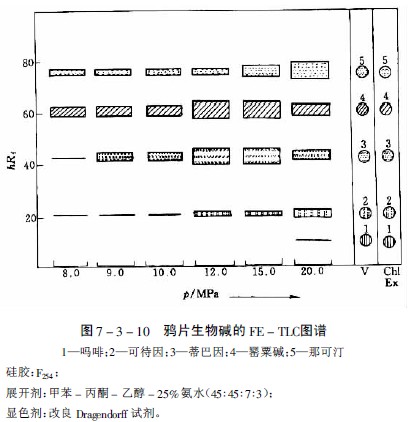
在研究蘿芙木、長春花、奎寧和等含27種生物堿的提取性時,選出氧化亞氮為適宜的提取氣體,但提取能力隨生物堿而異。如分離中主要生物堿時,在約8PMa下和那可汀很易提出,蒂巴因和可待因則較難提出,而嗎啡即使壓力達20MPa也僅提出極少量。提取一薄層色譜見圖7-3-10及圖7-3-11。
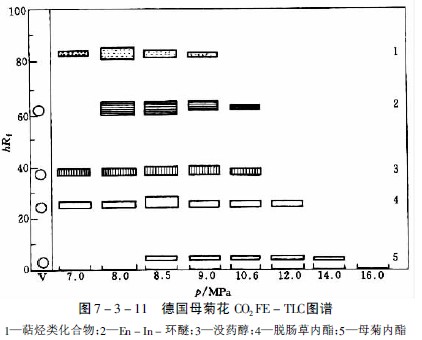
母菊內酯是德國母菊花的有效成分,極易受熱分解產生藍色的菊奧 ,因而無法用水蒸氣蒸餾獲得。用溶劑提取則得率低,如用二氧化碳超臨界流體提取,通過部分提取的方法,可得到母菊內酯及揮發性含氧化合物,經硅膠柱色譜分離即可得到母菊內酯結晶。
北京威鋒技術開發公司



免責聲明
- 凡本網注明“來源:化工儀器網”的所有作品,均為浙江興旺寶明通網絡有限公司-化工儀器網合法擁有版權或有權使用的作品,未經本網授權不得轉載、摘編或利用其它方式使用上述作品。已經本網授權使用作品的,應在授權范圍內使用,并注明“來源:化工儀器網”。違反上述聲明者,本網將追究其相關法律責任。
- 本網轉載并注明自其他來源(非化工儀器網)的作品,目的在于傳遞更多信息,并不代表本網贊同其觀點和對其真實性負責,不承擔此類作品侵權行為的直接責任及連帶責任。其他媒體、網站或個人從本網轉載時,必須保留本網注明的作品第一來源,并自負版權等法律責任。
- 如涉及作品內容、版權等問題,請在作品發表之日起一周內與本網聯系,否則視為放棄相關權利。


 采購中心
采購中心