


 8
8

【重要性】合規軟件只有審計追蹤和電子簽名還遠遠不夠
自2003年美國食品與藥品管理局出臺了聯邦法規第21章 第11款(FDA 21 CFR Part 11),對電子數據的真實性、完整性和可靠性,以及電子簽名有效性,收集和分析數據的軟件必須是經過驗證提出了要求。

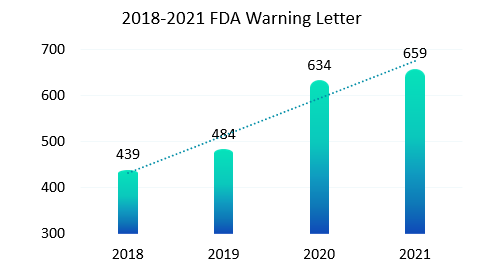
與紙質記錄相比,電子記錄是動態記錄必然會暴露出更多的不合規操作, 2015年警告信FDA 483表格和現場觀察報告頻頻出現,全球各地監管機構再次更新和完善了一輪法規要求。新法規施行后(2018版FDA 21CFR Part11),我們看到如下圖統計,最近幾年FDA發出的警告信也呈現出逐年上升趨勢。作為合規軟件的供應廠商,我們也明顯感受到國內客戶對于合規的要求也是越來越高。
每年年底/年初,制藥公司、生物技術公司、CRO、CDMO等都會進行合規的企業內部審計或國家審計局委派當地審計局進行審計。今年接到多次電話咨詢,無論內審還是外審都提到了操作員登錄軟件是否會被記錄到審計追蹤中。從審計關注點我們可以看出,國內對審計追蹤記錄完整性要求在提高,審計追蹤不僅僅記錄修改,還要記錄登錄/登出和查看信息;這也從側面反映法規對數據的隱私性越來越重視了。(可以放錢君娣上一篇關于審計追蹤的鏈接)。
Molecular Devices公司SoftMax Pro 7.1.2 GxP軟件除了能夠記錄完整的審計追蹤,還能夠記錄數據文件的生命周期,實現對數據來源和去向的全流程管理,真正做到無紙化、全流程電子記錄和電子簽名,最大限度保證數據記錄的真實性、完整性和可靠性。
為什么我們要對數據進行全流程管理?因為2020年12月1日正式施行的國家藥品監督管理局發布的藥品記錄與數據管理要求(試行)第二章基本要求中有如下要求,

針對這項要求SoftMax Pro 7.1.2 GxP軟件新增了數據文件狀態顯示(如下圖),在數據庫中和打開數據時都會顯示當前文件所處的生命周期狀態。
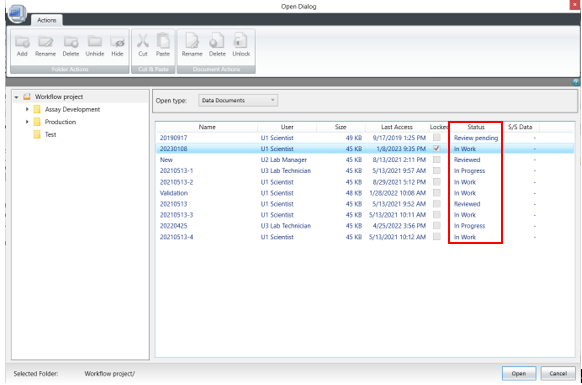
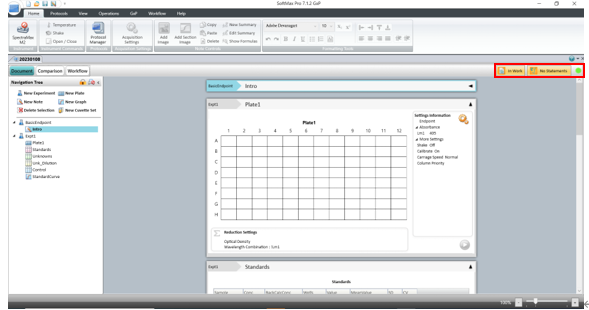
如Scientist權限新建數據狀態是in work表示正在進行模板方法開發,經過Lab Manager層層審批并電子簽名后可以更改文件狀態并將開發好的模板release出來,這時Lab Technician才有權限打開模板進行讀板和數據分析,簽名后數據將被鎖定不可更改,Lab Manager審核后電子簽名Approved。如果發現數據有問題,可以canceled;如果因為改進流程更新方法模板,可以將更改舊模板狀態成outdated,并不賦予Lab Technician打開outdated權限,以免使用錯誤模板讀板產生不合規的數據。
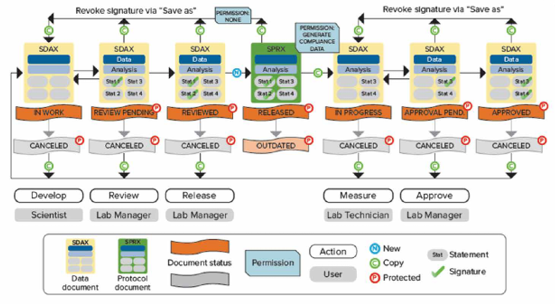
數據文件存儲在SQL Server數據庫中,文件的導入、重命名、移動、刪除、歸檔等都會記錄在審計追蹤中,真正做到了從數據來源到最終歸檔全流程管控。