化工儀器網
化工儀器網
公司動態
提供實驗數據分析服務,聚類分析,基因注釋數據分析
閱讀:1703 發布時間:2017-2-17世聯博研(北京)科技有限公司(BIO EXELLENCE INTERNATIONAL Tech Co.,Ltd)專注力學生物學(細胞組織生物分子力學)與3D生物打印,應廣大科研者要求,世聯博研在代理銷售科研儀器設備及配套耗材的同時提供細胞力學實驗技術服務和3D生物打印實驗技術服務以及數據分析服務。
世聯博研數據分析團隊由來自微軟、華為、中科院、農科院的力學、生物信息學、計算機專業人員組成,其中博士以上學歷者占50%以上,在圖像處理、蛋白質組學、轉錄組學、基因組學、數值模擬及數據可視化處理方面擁有豐富的經驗。公司建立了高性能計算平臺,具有強大的數據儲存和處理能力,使用Linux、R、Perl、Python、C++等工具進行數據處理,可為客戶定制數據分析服務并提供咨詢,將符合期刊發表要求的結果發送給客戶。
數據分析服務 世聯博研數據分析團隊由來自微軟、華為、中科院、農科院的力學、生物信息學、計算機專業人員組成,其中博士以上學歷者占50%以上,在圖像處理、蛋白質組學、轉錄組學、基因組學、數值模擬及數據可視化處理方面擁有豐富的經驗。公司建立了高性能計算平臺,具有強大的數據儲存和處理能力,使用Linux、R、Perl、Python、C++等工具進行數據處理,可為客戶定制數據分析服務并提供咨詢,將符合期刊發表要求的結果發送給客戶。
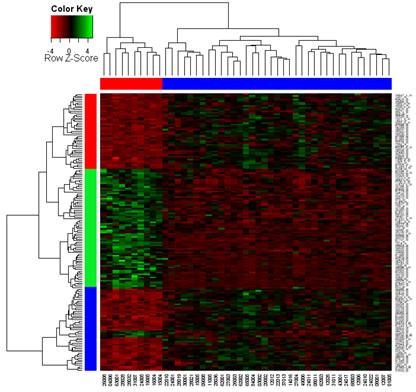
聚類分析(cluster analysis)是一類將數據所研究對象進行分類的統計方法。這一類方法的共同點是事先不知道類別的個數與結構;據以進行分析的數據是對象之間的相似性或相異性的數據。將這些相似(相異)性數據看成是對象之間的“距離”遠近的一種度量,將距離近的對象歸入一類,不同類之間的對象距離較遠。這就是聚類分析方法的共同思路。具體在生物學研究中,基因表達譜分析經常采用聚類分析的方法,其目的就是將基因或者樣本進行分組。從數學的角度,聚類得到基因分組,組內各成員在數學特征上彼此相似,但與其它組中的成員不同。其基本假設是組內基因的表達譜相似,它們可能具有功能相關性。大量功能相關的基因,特別是被共同的轉錄因子調控的基因表達譜非常相似,它們的產物可能構成蛋白質復合體,或者處于同一個調控通路中,因此還可以據此推測未知基因的功能并評估實驗的合理性(圖1)。 聚類分析根據分類對象不同分為Q型聚類和R型聚類。Q型聚類是指對樣本進行聚類,R型聚類是指對變量進行聚類分析。根據聚類方法可以分為系統聚類和動態聚類。系統聚類法一次形成類后就不再改變,而動態聚類開始先粗略地分一下類,然后按照某種*原則修改不合理的分類,直至類分得比較合理,如K-均值聚類等,適用于大樣本的Q型聚類分析。
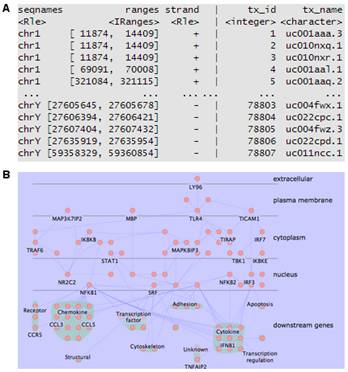
在基因芯片或者轉錄組學研究中,得到基因列表之后通常要對高達數千種基因或蛋白進行注釋,以得到其各種名字的對應關系、染色體定位及亞細胞定位來方便后續的研究。由于注釋數據庫的數量在不斷增加,且不斷進行著各種修改,所以在高通量組學研究中很難對這些信息進行整合。針對這些問題,我們開發了專門的組學數據注釋流程,可方便地進行基因ID轉換(圖2 A),并確定相應蛋白的亞細胞定位,以推測其功能(圖2 B)。
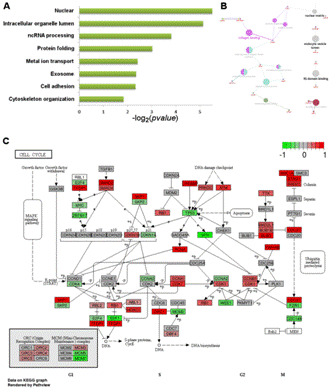
隨著轉錄組學及蛋白質組學的發展,現在已經可以一次性得到大量的基因表達數據。對這些數據進行分析時通常采用功能富集分析的方法(圖3),而非僅僅分析單個基因,以避免單基因分析可能產生的偏差,從而得到更準確的結論。進行功能富集分析需要可靠的數據庫和強健的算法(如累積超幾何分布、Fisher檢驗等),把涉及相同通路和功能的基因/蛋白質進行歸類,有助于生物學問題的解決。
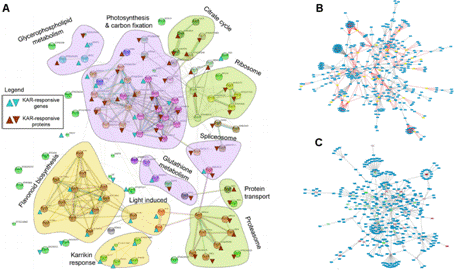
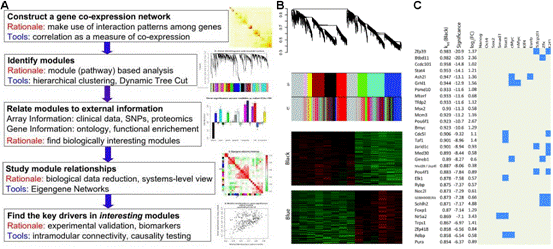
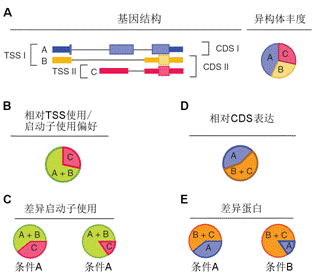
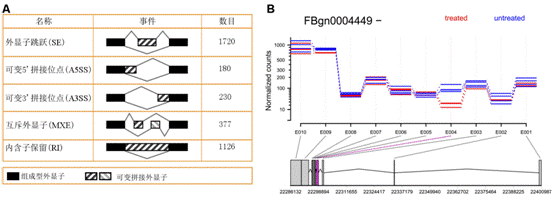
基因編碼的蛋白質不但會單獨行使功能,還會與其它蛋白質之間存在著相互作用,這種相互作用使其功能更加多樣化,且可以進行各種調控。所以,隨著后基因組時代的到來,蛋白質相互作用研究受到了越來越多的重視。現已有很多數據庫和工具進行蛋白互作(包括物理互作和功能互作)數據的儲存和處理,其數據主要來自于基因組結構、高通量實驗、共表達實驗和文獻挖掘。將蛋白質相互作用網絡進行圖形化展示,為其功能關系提供了高層次神力,有助于生物學過程的模塊化分析(圖4)。 5.權重基因共表達分析 常規的差異表達分析方法大大促進了生物學的發展,取得了很多重大發現,但是,這些方法都忽略了基因表達模式之間的相關性。結果,這些數據產生的信息數量很多,卻很難從中發現有價值的線索,無法確定差異表達基因的優先級,更難以去研究潛在的生物學通路。相反,相關性網絡(又稱為共表達網絡)可以發現彼此相關的基因(圖5 A),并將其分為相應的cluster(即共表達模塊)(圖5 B),然后計算得到模塊中權重zui高的基因,將其做為關鍵調節因子(圖5 C),從而簡化了數據的分析過程,能夠的從數據中提取出關鍵信息,現已有大量的研究采用了這種方法。 6.高能量測序數據分析(差異基因表達、差異異構體表達、可變拼接) 細胞內基因表達水平時刻處于變化之中,具有顯著的時間、組織、條件特異性,同時許多基因還具有不同的異構體(圖6.1),測定不同刺激條件下的基因及其異構體的表達變化對于闡明相關的生物學過程極為重要。RNAseq技術可以一次性鑒定出大量的差異表達基因/異構體,從而在系統水平了解生命活動的機制,也可以篩選出重要基因進行更深的功能研究。 可變拼接(AS)是真核生物基因表達調控的重要機制之一。RNAseq已成為定量分析細胞內的可變拼接的強有力工具,隨著高通量測序儀的不斷涌現,RNSseq的數據量也在以指數形式增加。在此背景下,我們提供了可變拼接分析服務,對特定基因以及大規模轉錄組數據的可變拼接(圖6.2 A)、差異外顯子使用(圖6.2 B)等進行定量分析。
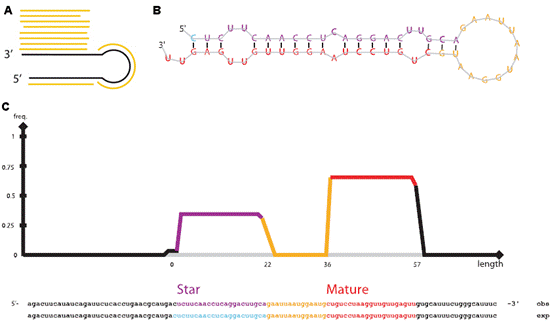
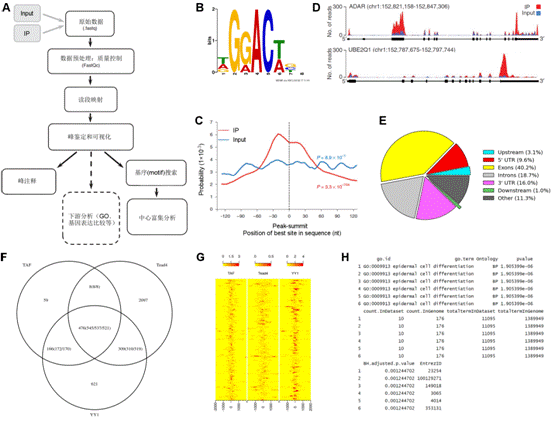
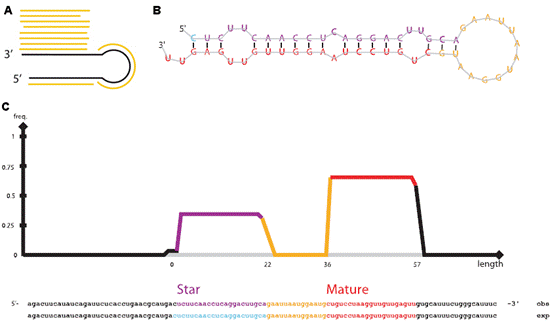
7.microRNA數據分析 MicroRNA (miRNA) 是一類由內源發卡結構轉錄本產生的長度約為22個核苷酸的非編碼單鏈RNA 分子,通過與靶mRNA分子互補配對進行轉錄后調控。提取細胞內全部RNA后進行小RNA建庫,然后進行高通量測序,通過特定的算法(圖7 A),由測序數據可得到已知的和新的miRNA分子前體(圖7 B),并對此前體產生的miRNA進行定量(圖7 C)。 8.染色質免疫共沉淀(ChIP)分析 染色質免疫沉淀結合高通量測序技術(ChIP-seq)是鑒定基因組范圍內DNA/RNA結合蛋白靶位點的標準方法,現已開始在力學生物學中得到應用,用于研究力學刺激下的蛋白質-DNA相互作用。Chip-seq先富集目標蛋白結合的DNA/RNA片段,然后純化和建庫并進行高通量測序。得到的原始數據經過特定的數據處理流程(圖8 A),可得到全基因組范圍內與目標蛋白互作的DNA序列信息(圖8 B、C)、基因不同位置的分布(圖8 D、E)、比較不同的生物學重復之間的重復性(圖8 F)、結合位點熱圖(圖8 G),并對峰相關的基因進行GO功能富集分析(圖8 H)等。 9、主成分分析 10、HITS-CLIP分析 11、宏基因組分析 12、外顯子測序分析 13、單細胞測序分析 |